Pātiki and Waharua Kōpito patterns
On this page
Key information
|
Mode of transmission |
Contact with infected blood or body fluids during childbirth (vertical transmission); sexual intercourse, intravenous drug use or contact with broken skin (horizontal transmission). |
|---|---|
|
Incubation period |
45–180 days, commonly 60–90 days. |
|
Period of communicability |
Potentially infectious 2–3 weeks before the onset of symptoms, during the clinical disease and usually for 2–3 months after acute hepatitis B illness; or for as long as HBsAg continues to be present in blood (chronic hepatitis B carrier state). |
|
Incidence and burden of disease |
New Zealand is a country with a low overall prevalence of hepatitis B carriage, but it contains certain populations with high prevalence. All pregnant women and high-risk groups should be screened for chronic HBV infection. HBV acquisition in infancy is very likely to lead to chronic infection. Chronic HBV infection can progress to cirrhosis and liver cancer. |
|
Funded vaccines |
HepB (Engerix-B) DTaP-IPV-HepB/Hib (Infanrix-hexa). |
|
Dose, presentation, route |
HepB:
DTaP-IPV-HepB/Hib:
|
|
Funded vaccine indications and schedule |
At ages 6 weeks, 3 months and 5 months: DTaP‑IPV‑HepB/Hib. Infants born to HBsAg-positive mothers should receive HepB vaccine plus HBIG at birth, then the usual childhood schedule. Serological testing at age 9 months (anti-HBs and HBsAg). Individuals with eligible conditions or close household contacts of infected individuals: HepB (see section 9.5). |
|
Recommended, unfunded |
Those with increased risk from occupational or sexual exposure to body fluids and faeces, or receiving regular blood products. Those with developmental disability, current or prior injectable drug users, prison inmates, and travellers to and from high-prevalence countries. |
|
Vaccine effectiveness |
In general, efficacy is 85–95 percent in high-risk groups, though likely to be lower in older individuals and those with immunocompromise. Protection is expected to be lifelong and boosters are not required. |
|
Public health measures |
Notify all cases of acute hepatitis B infection (see section 9.8). |
|
Post-exposure prophylaxis |
Vaccination and immunoglobulin prophylaxis of close contacts, as appropriate (see section 9.8). |
9.1. Virology
The hepatitis B virus (HBV) is a partially double-stranded DNA virus belonging to the Hepadnaviridae family. Three major subunits make up the structural components:
- the HBV genome, a small, circular, partially double-stranded DNA molecule, in association with a polymerase enzyme
- the nucleocapsid core, which surrounds the genome and consists of core protein (hepatitis B core antigen, HBcAg)
- the outer lipoprotein envelope, which contains the hepatitis B surface antigen (HBsAg).
The genome has four genes (S, C, X and P). Both the core nucleocapsid protein (HBcAg) and the ‘early’ protein (which makes HBeAg) are translated from the C gene. HBcAg is essential for viral packaging and is an integral part of the nucleocapsid. HBeAg is a soluble protein that is not part of the virus particle. Detection of HBeAg in the serum is correlated with viral replication and is a marker for severe disease. It is most commonly found in those with acute hepatitis B and those with chronic HBV infection with high viral load.[1]
9.2. Clinical features
There is a broad spectrum of clinical disease with HBV infection, from subclinical through to fulminant hepatitis. Persistent infection can lead to chronic liver disease, potentially causing cirrhosis or hepatocellular carcinoma.
9.2.1. Serological markers of infection
9.2.1. Serological markers of infection
The HBV antigens and their associated antibodies are serological markers of HBV infection or vaccination (Table 9.1). At least one serological marker is present during the different phases of infection (Table 9.2).
Table 9.1: HBV antigens and their respective antibodies
|
Antigen |
Antibody |
|---|---|
|
HBsAg |
Anti-HBs |
|
HBcAg |
Anti-HBc |
|
HBeAg |
Anti-HBe |
Table 9.2: Interpretation of serology for HBV infection
|
Serological marker |
Interpretation |
|||
|---|---|---|---|---|
|
HBsAg |
Total |
IgM |
Anti-HBs |
|
|
– |
– |
– |
– |
Never infected |
|
+ |
– |
– |
– |
Early acute infection; transient (up to 18 days) after vaccination |
|
+ |
+ |
+ |
– |
Acute infection |
|
– |
+ |
+ |
+ or – |
Acute resolving infection |
|
– |
+ |
– |
+ |
Recovered from past infection and is immune |
|
+ |
+ |
– |
– |
Chronic infectiona |
|
– |
– |
– |
+ |
Immune if ≥10 IU/L vaccinated or natural infection |
|
Key: Anti-HBc = antibody to hepatitis B core antigen; anti-HBs = antibody to hepatitis B surface antigen (HBsAg); IgM = immunoglobulin M; + = positive test result; – = negative test result.
a. HBeAg positive (HBeAg+) correlates with high viral load and increased risk of transmission; HBeAg negative (HBeAg–) correlates with lower viral load and reduced risk of developing cirrhosis or cancer.
Adapted from: Van Damme P, Ward J, Shouval D, et al. 2018. Hepatitis B vaccines. In: Plotkin SA, Orenstein WA, Offit PA (eds). Plotkin’s Vaccines (7th edition). Philadelphia, US: Elsevier. Table 25.1. |
||||
Any difficulties with interpreting serological results for cases and contacts should be discussed with an infectious diseases physician or the laboratory. See the ‘Hepatitis B’ chapter of the Communicable Disease Control Manual for recommendations for HBV case and contact management.
9.2.2. Acute hepatitis
9.2.2. Acute hepatitis
The virus preferentially infects liver cells, multiplying in the liver and releasing large amounts of HBsAg, which is present in the blood of people with active infection. The incubation period varies between 45 and 180 days and is commonly 60 to 90 days.
HBV is not directly cytopathic; the host’s immune response leads to death of infected liver cells. Most infected people mount an effective immune response that leads to eradication of infection over a period of several months. Approximately 80 percent of adults with acute infection have symptomatic hepatitis, and the remaining 20 percent can be asymptomatic (but these proportions vary).[2]
The common symptoms of acute hepatitis B illness are fever, jaundice, malaise, anorexia, nausea, vomiting, myalgia and abdominal pain. Jaundice usually develops within two weeks of onset of the illness, and dark urine and/or clay coloured stools might appear up to five days before clinical jaundice. Clinical signs and symptoms of acute hepatitis B usually resolve one to three months later.[1]
There is a small risk of liver failure (less than 1 percent) with acute infection; if failure occurs, almost half will die or require emergency liver transplantation.
9.2.3. Chronic HBV infection
9.2.3. Chronic HBV infection
The main burden of HBV disease occurs in people with chronic HBV infection. Chronically infected people are identified by presence and persistence of HBsAg in their serum for at least six months. The age of acquisition of HBV is strongly associated with the risk of developing chronic HBV infection. Approximately 90 percent of those infected perinatally or in infancy develop chronic HBV infection, compared with 30 percent of children infected between ages 1 and 4 years and less than 5 percent of people infected as adults.
Infants seldom mount an immune response to HBV infection, and infection in infancy is often asymptomatic. Asymptomatic chronic infection stimulates persistent immune responses that may eventually lead to cirrhosis (decades later); cirrhosis and chronic infection increase the risk of development of hepatocellular carcinoma.
Table 9.3: Characteristics and phases of chronic hepatitis B virus infection
|
Phases of HBV infection |
Features |
|---|---|
|
Immune tolerance phase |
Prolonged period of active viral replication without active liver disease. Seen in children who acquire infection perinatally. |
|
Immune clearance phase |
Active viral replication and active liver disease |
|
Inactive chronic HBV infection |
Low or absent viral replication and remission of active liver disease |
|
Reactivation |
HBV replication after inactivity, seen in some patients |
There are up to four phases of chronic infection as in Table 9.3 show; not all are present in all infections.[1] The initial phase of infection may last 10 to 30 years, during which spontaneous clearance rates of HBeAg in the serum are less than 1 percent per year.
Chronically infected people who are HBsAg positive can also have detectable HBeAg in the serum; this combination is considered most infectious. Although recent evidence suggests HBeAg negative patients are less infectious, it is dependent on HBV DNA levels. Whatever the case, both groups can be an ongoing source of infection to susceptible individuals. In the early years of chronic infection, high rates of viral replication are common, and both HBeAg and high levels of HBV DNA are present in the blood. In later years, HBeAg may be absent from the blood, and HBV DNA levels (viral load) are usually lower, both of which correspond with lower rates of viral replication.
It is estimated that 4.5 percent of HBsAg positive individuals (12 million people worldwide) have been co-infected with hepatitis D virus (HDV), which is a significant contributor to HBV-associated cirrhosis and hepatocellular carcinoma.[3] The highest prevalence is seen in those with hepatitis C and HIV, and in certain geographic areas.
9.2.4. Routes of transmission
9.2.4. Routes of transmission
HBV is usually transmitted through contact with infected blood or body fluids during childbirth, contact with broken skin, sexual intercourse or intravenous drug use. Although HBV can be found in all body fluids, blood has the highest concentration and saliva the lowest. HBV in dried blood remains infective for at least one week.[4]
Perinatal (vertical) transmission
The primary source of HBV infection is perinatal exposure from mothers with chronic HBV infection. Transmission usually occurs at the time of birth. The in utero transmission of HBV is relatively rare, accounting for less than 2 percent of infections transmitted from mother to infant.[5]
If no prophylaxis is given to the infant, the baby of an HBeAg positive mother has a 70–90 percent risk of infection, while the baby of an HBeAg negative HBsAg positive carrier mother has a 5–20 percent risk of infection. Over 90 percent of infants who acquire infection perinatally become chronic carriers.
Person-to-person (horizontal) transmission
Non-sexual person-to-person transmission probably occurs from inadvertent percutaneous or mucosal contact with blood or infectious body fluids among people in close daily contact (household members).
The main sources of transmission are:
- sexual contact with an infected individual
- percutaneous exposure to blood or infectious body fluids
- needle-stick injuries or sharing needles.
Those travelling to high endemic countries are at higher risk of exposure (see below).
9.3. Epidemiology
9.3.1. Global burden of disease
9.3.1. Global burden of disease
Approximately two billion people worldwide had been exposed to HBV in 1995. In 2015, based on serological data, around 3.5 percent of the general population globally were infected with HBV and more than 250 million people were estimated to have chronic infection and these people remain at risk of developing cirrhosis and hepatocellular carcinoma.[6, 7] More than 90 percent of individuals with chronic HBV resided in the Asia–Pacific region, where most countries have high prevalence rates of HBV infection (the population rate of HBsAg positivity is between 5 and 20 percent) and more than 99 percent of HBV-infected people in this region acquired infection through vertical transmission from their mother (usually at the time of delivery) or in early childhood.[8] As an example of this risk, 22.8 million out of 80 million people living in China with chronic HBV infection are women of child-bearing age.[9] Acquisition of HBV during adulthood (usually via sexual transmission or injecting drug use) is associated with a high rate of symptomatic hepatitis but a low rate of chronic infection.
The introduction of universal childhood HBV immunisation has changed the epidemiology of chronic infection in many countries, but it will be several decades (one to two human generations) before the full benefits are realised. In China, for example, within 20 years since the introduction of HBV immunisation, mother-to-child transmission has been cut by 97 percent; 120 million new HBV infections and 28 million chronic infections have been averted.[9] Thirty years after the introduction of a HepB immunisation programme for newborns in Taiwan, infant fulminant hepatitis mortality and, in those aged 5 to 29 years, chronic liver disease and hepatocellular carcinoma mortality had all decreased by more than 90 percent.[10]
The world can be divided into regions with high (8 percent and over), high-moderate (5–7 percent), low-moderate (2–4 percent) and low (less than 2 percent) prevalence of chronic infection, defined as the presence of HBsAg in serum.[11, 12] In regions with a high prevalence of chronic infection, the lifetime risk of exposure to HBV is almost 80 percent, with most infections occurring in the first decade of life. The Pacific Islands and most of Asia (except Japan and India) are high-prevalence regions. Other high-prevalence regions include Sub-Saharan Africa and Latin America.[12] In contrast, in countries with a low HBsAg prevalence, the lifetime risk of HBV exposure is less than 20 percent, with most infections acquired in adulthood. New Zealand has a low overall prevalence of hepatitis B carriage but contains certain populations with high prevalence (see section 9.3.2 below).
9.3.2. New Zealand epidemiology
9.3.2. New Zealand epidemiology
Before the introduction of HBV immunisation in New Zealand, HBV transmission was common among preschool and school-aged children. The exact mode of transmission is uncertain, but is thought to be related to close contact. In the eastern Bay of Plenty region almost half of the population were infected by age 15 years.[13, 14] Even after the introduction of universal HepB in 1988 (see Appendix 1), there were regions in New Zealand where children were still at risk of HBV infection due to poor immunisation coverage rates.[15, 16, 17]
Acute HBV infection
Only acute hepatitis B is a notifiable disease in New Zealand; notification rates do not describe the burden of chronic HBV infections.
The HBV notification rate in 2019 was 0.6 per 100,000 population (28 cases), similar to the 2018 rate (0.7 per 100,000, 33 cases). The highest notification rate was in the 30–39 years age group (1.1 per 100,000) followed by 50–59 years and 70 years and over (both 0.8 per 100,000). The notification rate was higher for males (0.9 per 100,000) than for females (0.3 per 100,000) (ESR, 8 June 2020).
Ethnicity was recorded for all cases. The Māori (1.3 per 100,000) ethnic group had the highest hepatitis B notification rate followed by the Asian (0.7 per 100,000) ethnic group.
The most common reported risk factors were overseas travel, migration and sexual contact with a confirmed case or carrier.
Hepatitis B notifications have declined from 609 cases in 1984 to 28 cases in 2019 (see Figure 9.1). While difficult to quantify accurately, the introduction of universal infant immunisation in 1988 has contributed to the dramatic decline in the number of newly notified cases of HBV infection.
Figure 9.1: Notifications of hepatitis B, 1997–2019
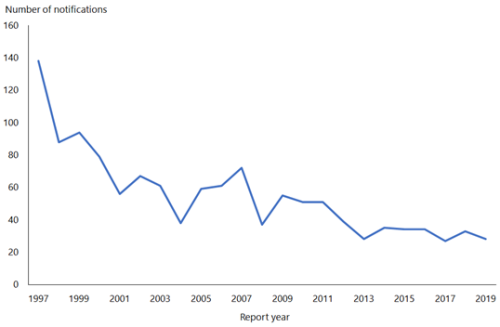
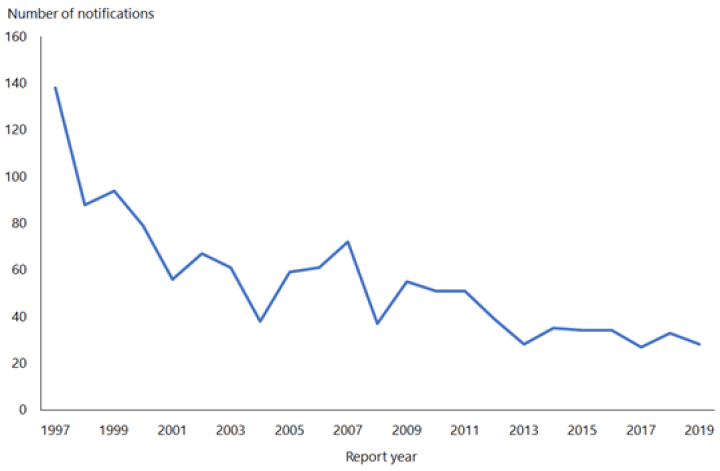
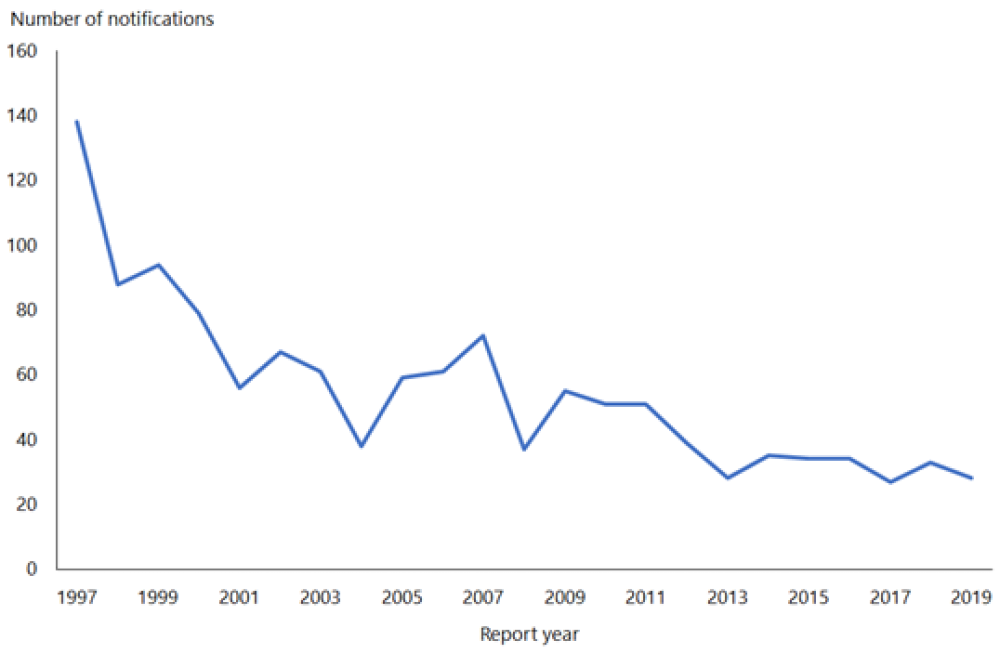
Source: ESR
For recent data on acute hepatitis B notifications, refer to the most recent notifiable disease annual reports from PHF Science (formerly ESR) (external link).
Chronic HBV infection
The Hepatitis Foundation of New Zealand reports that around 120,000 people in New Zealand are living with chronic HBV infection, around 50 percent are diagnosed, but only around 7,000 are being treated. Based on 2016 data, around 1,000 new cases are diagnosed each year nationally. Mathematical modelling anticipated that the prevalence of chronic HBV infection would drop from 3.3 percent in 2016 to 2.4 percent by 2030, falling short of the elimination targets set by the WHO in 2016.[18]
The National Hepatitis B Screening Programme found that between 1999 and 2002 in the North Island, the highest rates of chronic HBV infection were among Chinese (9.1 percent), Pacific peoples (8.5 percent) and Māori (5.8 percent). Although Europeans were not specifically targeted in this screening programme, they have an estimated prevalence rate of 1 percent (higher than in Australia, North America and Europe), reflecting an increased risk of childhood horizontal transmission.[19]
A New Zealand-based modelling study estimated that until the year 2100, people with chronic HBV infection will continue to provide a source of infection to susceptible people.[20] Increased immigration from high-prevalence countries in the Asia–Pacific region is also likely to influence HBV prevalence in New Zealand.
Because people who acquire chronic HBV infection in childhood usually do not develop hepatocellular carcinoma until aged 40 years or older, the introduction of a universal HBV vaccination in 1988 is unlikely to have a significant effect on the incidence of hepatocellular carcinoma until approximately 2030.
A retrospective laboratory data study of antenatal HBsAg tests from the Midlands region (Bay of Plenty, Eastern Bay of Plenty, Waikato and Rotorua) between 1997 and 2009 found a declining prevalence of HBV infection. This decrease was seen across all age groups, but was most marked in antenatal tests of women aged under 20 years, due to receipt of funded HepB in childhood.[21]
A long-term follow-up study in New Zealand showed that horizontally acquired HBV infection during childhood in Māori and Pacific peoples correlates with increased rates of hepatocellular carcinoma and liver-related mortality.[22] This study emphasises the importance of early protection of the infant with vaccination.
Strategy for prevention
In 1988 New Zealand was one of the first countries to introduce universal infant hepatitis B immunisation. As of 31 December 2019, 93 percent of New Zealand children aged 2 years had completed a primary course of HepB, which confers lifelong immunity in approximately 95 percent of those vaccinated.
9.4. Vaccines
9.4.1. Available vaccines
9.4.1. Available vaccines
The specific monovalent and combination HepB vaccines licensed (approved for use) and available (marketed) in New Zealand contain recombinant HBsAg (HepB).
Funded vaccines
- HepB (Engerix-B, GSK): contains 20 µg HBsAg per 1 mL dose (10 µg HBsAg per 0.5 mL paediatric dose); it does not contain a preservative. Other components and residuals include aluminium hydroxide, sodium chloride, sodium phosphate dehydrate, sodium dihydrogen phosphate and traces of polysorbate 80.
- DTaP-IPV-HepB/Hib (Infanrix-hexa, GSK): diphtheria, tetanus, acellular pertussis, inactivated polio, hepatitis B and Haemophilus influenzae type b vaccine (see section 6.4.1 for more information).
Other vaccines
HepA-HepB (hepatitis A and hepatitis B vaccine): Twinrix and Twinrix Junior (GSK) (see also section 8.4.1).
9.4.2. Efficacy and effectiveness
9.4.2. Efficacy and effectiveness
Clinical trials in high-risk groups have shown a vaccine efficacy of 85–95 percent for HepB vaccines.[8]
See also section 16.4.2 for information about the DTaP-IPV-HepB/Hib vaccine.
Immunogenicity
Serum anti-HBs antibody ≥10 IU/L, measured 1–2 months after immunisation, is considered by WHO as a correlate of long-term protection.[8] In the primary care setting, individuals who have had a documented seroconversion after three injections are expected to have lifelong immunity with no need for further boosters, even if circulating antibody is subsequently not detectable.
Smoking, obesity, HIV infection and chronic disease (including renal failure) all reduce vaccine efficacy, but age is the primary factor affecting the response. At least 98 percent of infants, 95 percent of children and 90 percent of adolescents develop protective levels of antibody after three doses of vaccine. Some non-responders will not produce adequate antibody levels to the initial vaccination course, but most respond to further vaccine doses.
However, some people are persistent non-responders. Persistent non-responders often have an impaired immune system, such as organ transplant recipients and those with HIV infection or chronic disease, including advanced cirrhosis, renal failure or those undergoing haemodialysis. A small percentage (approximately 2–3 percent) of the immunocompetent population may also fail to elicit an antibody response. High-risk individuals who fail to respond adequately are recommended further vaccinations (see section 9.5.7).
Effectiveness of birth dose given to babies born to HBsAg‑positive mothers
Infants vaccinated at birth born to infected mothers were 3.5 times less likely to be infected with HBV than those who did not receive a birth vaccination.[8, 23] For babies of HBeAg-positive mothers, controlled trials have shown that vaccine at birth provides 75 percent protection from infection, while administration of HBIG in addition to vaccination provides 85–95 percent protection against transmission.[23, 24] Protection is reduced to less than 80 percent when the mother’s HBV DNA level (viral load) is greater than 108 IU/mL (or 108 copies/mL).[25] In this situation, administration of tenofovir (an antiviral agent) to the mother during the last trimester is recommended and funded.
Duration of immunity
The development of anti-HBs antibodies after a primary vaccination course (three injections and seroconversion) indicates development of immune memory. The quantity of antibody in serum is thought to determine the length of time the antibody titre can be detected in the blood, although any reading ≥10 IU/L post-vaccination course is considered protective.[26, 27] Children who are given booster doses up to 12 years after the primary series show strong anamnestic (secondary) responses, indicating that booster is unnecessary once a seroprotective level is reached after the three-dose primary vaccination course.[26, 27]
Long-term protection from clinical infection, despite loss of detectable neutralising antibody, is thought to reflect a strong cellular memory immune response following HBV vaccination.[28] Even though a large proportion of vaccine recipients may have undetectable antibody within seven years of vaccination, there is evidence from Germany,[28] Taiwan,[29] Alaska[30] and Hawaii[31] that boosters of HepB are unnecessary following completion of infant immunisation.
Sustained immune memory, including circulating memory B and T cells, and long-term protection have been shown 20–30 years after complete primary immunisation of immune competent adults in the absence of natural or artificial boosting.[24]
In general, vaccine recipients who are subsequently infected with HBV do not develop clinical illness but may have anti-HBc present in plasma.[1]
Impact on chronic HBV infection
In all populations, where it has been measured, immunisation has led to a dramatic drop in HBV chronic infection.[32] For example, chronic HBV infection dropped from 16 percent to zero in Alaska as a result of 96 percent immunisation coverage. In Taiwan, the incidence of hepatocellular carcinoma also decreased as a result of the immunisation programme in children.[33, 34] Adolescents and adults who were offered universal HBV vaccination in infancy had more than 75 percent lower prevalence of HBV infection and anti-HBc prevalence than those for whom immunisation was unavailable.[35]
9.4.3. Transport, storage and handling
9.4.3. Transport, storage and handling
Transport according to the National Standards for Vaccine Storage and Transportation for Immunisation Providers 2017 (2nd edition).
Store at +2°C to +8°C. Do not freeze. DTaP-IPV-HepB/Hib and HepB vaccines should be stored in the dark.
DTaP-IPV-HepB/Hib (Infanrix-hexa) must be reconstituted by adding the entire contents of the supplied container of the DTaP‑IPV-HepB vaccine to the vial containing the Hib-PRP pellet. After adding the vaccine to the pellet, the mixture should be shaken until the pellet is completely dissolved. Use the reconstituted vaccine as soon as possible. If storage is necessary, the reconstituted vaccine may be kept for up to eight hours at 21°C.
9.4.4. Dosage and administration
9.4.4. Dosage and administration
DTaP-IPV-HepB/Hib
Each 0.5 mL dose of DTaP-IPV-HepB/Hib (Infanrix-hexa) vaccine contains 10 μg of HBsAg, and is administered by intramuscular injection (see section 2.2.3).
HepB
The dose of HepB vaccine varies according to the vaccine manufacturer, the age of the individual and/or their health status (see section 9.5 for recommendations):
- Engerix-B 20 μg (GSK): 20 μg HBsAg per 1.0 mL
- Engerix-B paediatric 10 μg (GSK): 10 μg per 0.5 mL.
HepB vaccine is administered by intramuscular injection. It can be also administered by subcutaneous injection, if indicated for bleeding disorders (see section 2.2.3).
Co-administration with other vaccines
Hepatitis B vaccines may be given at the same time as all other vaccines on the Schedule, including measles, mumps and rubella (MMR) vaccine.
If a course of vaccine is interrupted, it may be resumed without repeating prior doses (see Appendix 2).
9.5. Recommended immunisation schedule
Table 9.4: Hepatitis B vaccine recommendations, funded and unfunded
Table 9.4: Hepatitis B vaccine recommendations, funded and unfunded
| Note: Funded individuals and situations are in the shaded rows. See the Pharmaceutical Schedule (external link) for any changes to funding decisions. |
|
Recommended and funded |
|---|
|
Household or sexual contacts of HBsAg-positive patients (ie, patients with acute or chronic HBV infection) |
|
Babies of HBsAg-positive mothers (ie, mothers with acute or chronic HBV infection) – require a birth dose plus the three-dose primary series (HBIG is also given to these babies at birth) |
|
Children and adolescents aged under 18 years who are considered not to have achieved a positive serology by 1 month after vaccination and require additional vaccination or require a primary course of vaccinationa |
|
Individuals who are HIV-positiveb |
|
Individuals who are hepatitis C-positivec |
|
Following non-consensual sexual intercourse |
|
Prior to planned or following immunosuppressionb,d |
|
Prior to or following solid organ transplantb,d |
|
Individuals post-HSCTb |
|
Following needle-stick injury |
|
Patients on dialysisb,d |
|
Recommended, not funded |
|
Adults at occupational risk (see section 4.8) |
|
Adults at risk of infection by sexual exposure:
|
|
Individuals with haemophilia and other regular recipients of blood products |
|
Prison inmates |
|
Current or recent injecting drug users |
|
Individuals with developmental disabilities |
|
Migrants from HBV endemic countries (HBsAg prevalence ≥2 percent)f |
|
Travellers to HBV endemic regions (HBsAg prevalence ≥2 percent)f |
|
a. Serological testing is not routinely recommended, see Figure 9.3. b. See also section 4.3.2. c. Hepatitis C patients should also receive hepatitis A vaccine, although this is not currently funded. d. The period of immunosuppression due to steroid or other immunosuppressive therapy must be longer than 28 days. e. 40 µg of HepB is recommended for adult dialysis patients or for adult liver or kidney transplant patients.[36] See Table 9.5. f. See the Centers for Disease Control and Prevention website (external link) for countries with an HBsAg prevalence ≥2 percent. Consider combined Hep A and B vaccination for travellers to these regions. |
9.5.1. Usual childhood schedule
9.5.1. Usual childhood schedule
A primary course of hepatitis B vaccination is given as three doses of DTaP-IPV-HepB/Hib at ages 6 weeks, 3 months and 5 months (Table 9.5). If a course of immunisation is interrupted for any reason, it may be resumed without repeating prior doses (see section 9.5.3 and Appendix 2).
Table 9.5: Usual childhood schedule for hepatitis B-containing vaccine (excluding catch-up)
|
Age |
Vaccine |
Comment |
|---|---|---|
|
6 weeks |
DTaP-IPV-HepB/Hib |
Primary series |
|
3 months |
DTaP-IPV-HepB/Hib |
Primary series |
|
5 months |
DTaP-IPV-HepB/Hib |
Primary series |
Preterm infants of HBsAg-negative women
Some low birthweight or preterm infants may have a reduced response to HepB vaccine at birth.[37] However, by the chronological age of 1 month, all medically stable preterm infants, regardless of initial birthweight or gestational age, respond to HepB as well as term and larger infants.[38] Because New Zealand’s Schedule starts at age 6 weeks, low birthweight and preterm infants are expected to respond to HepB content in the DTaP-IPV-HepB/Hib vaccine. (See also sections 4.2.1 and 4.2.2.)
Infants with liver or renal disease
HepB vaccine is funded for liver or kidney transplant patients and for dialysis patients. For infants requiring transplants, see ‘Solid organ transplantation’ in section 4.3.10. For infants undergoing dialysis, see ‘Chronic kidney disease’ in section 4.4.
9.5.2. Infants born to HBsAg-positive mothers
9.5.2. Infants born to HBsAg-positive mothers
The routine schedule for these infants is a birth dose of monovalent HepB plus HBIG, then three routine doses of DTaP-IPV-HepB/Hib at ages 6 weeks, 3 months and 5 months.
All pregnant women should receive antenatal screening for hepatitis B infection by testing for HBsAg. Infants of HBsAg-positive mothers are to be notified at birth using the form HE1446: Consent for hepatitis B vaccine and hepatitis B immunoglobulin and notification to the Medical Officer of Health (external link), available from the Health Ed website (external link) or the local authorised health education resource provider or public health unit.
Infants born to HBsAg-positive mothers should receive:
- 100–110 IU HBIG neonatal, at or as close as possible to birth
- a birth dose of HepB which should be given at or as close as possible to birth (preferably within 12 hours).
If HBIG and/or HepB is inadvertently omitted, administer as soon as the omission is recognised. HBIG can be administered up to seven days post-delivery. If there is a delay for longer than seven days, seek specialist advice.
These infants should then continue as per the Schedule at ages 6 weeks, 3 months and 5 months. Serological testing is required at 9 months of age (see below).
The vitamin K injection may also be given at the same time, in the same limb as the HBIG, but not at the same site.
Occasionally women have not been tested for their HBsAg status during the antenatal period. If a woman’s HBsAg status is unknown at the time of delivery, the baby should be given HepB at the time of delivery while waiting for the result of an urgent HBsAg test on the mother. If she is found to be HBsAg positive, the baby should be given HBIG as soon as possible, up to seven days post-delivery. Immunoprophylaxis is most effective when given within 12 hours of delivery.[38] Subsequent vaccine doses are given as per the Schedule.
It is essential to take blood to determine whether the baby has seroconverted (anti-HBs positive) or has become infected despite immunoprophylaxis (HBsAg positive), or is neither infected nor immune (ie, HBsAg negative and anti-HBs negative). Testing should be performed at 9 months of age to avoid detection of anti-HBs from HBIG administered during infancy and to maximise the likelihood of detecting late onset HBV infections.[38] Infants of HBsAg-positive mothers should be placed on a practice recall system to have their blood tested at 9 months of age. Check at the 12-month immunisation event to ensure that testing has occurred. The serology results should be interpreted as in Figure 9.2.
Neonatal HBIG plus vaccine will fail to prevent vertical HBV transmission in up to 20 percent of infants born to HBsAg-positive mothers with serum HBV DNA levels greater than 108 IU/mL (or 2 x107 copies/mL). These mothers are usually young, with normal alanine transaminase, and are HBeAg-positive. If the mother’s HBV DNA level is greater than 200,000 IU/mL[39, 40, 41], administration of tenofovir (an antiviral agent) during the last trimester is funded.
The number of such high-risk pregnancies appears to be increasing in this country as a result of the immigration of young Asian women of childbearing age, of whom approximately 8 percent are HBsAg-positive with the majority of those also HBeAg-positive. In contrast, the number of HBsAg-positive Māori and Pacific women of childbearing age has decreased markedly due to infant vaccination. In addition, most HBsAg‑positive Māori and Pacific women are HBeAg-negative, with lower HBV DNA levels (below 108 IU/mL).
Infants born to mothers who received oral antiviral therapy for chronic HBV must still receive the recommended neonatal HBIG/vaccine schedule. All other vaccines are administered as per the Schedule.
See Appendix 6 and section 9.8.1 for more information about passive immunisation and HBIG.
Preterm and low birthweight infants of HBsAg-positive women
Preterm and low birthweight infants of HBsAg-positive women should be managed as above, regardless of birthweight or gestation.
Figure 9.2: Management of an infant of an HBsAg-positive woman
Figure 9.2: Management of an infant of an HBsAg-positive woman
|
Screen all women in early pregnancy for hepatitis B carriage |
|
|---|---|
|
Woman is HBsAg positive No → See section 9.5.1: ‘Usual childhood schedule’ |
|
|
Yes ↓ |
|
|
All HBsAg-positive pregnant women should also be tested for HBeAg and should have HBV DNA measured. The results should be discussed with a specialist or the woman should immediately be referred to a specialist for ongoing care. Give the baby hepatitis B protection as follows. |
|
|
At age |
Action to be taken |
|
Birth |
Give HBIG 100–110 IU and HepB |
|
6 weeks |
DTaP-IPV-HepB/Hib |
|
3 months |
DTaP-IPV-HepB/Hib |
|
5 months |
DTaP-IPV-HepB/Hib |
|
9 months |
Take a blood test to check for hepatitis B infection (HBsAg) and for vaccine-induced immunity (anti-HBs). If HBsAg is negative and anti-HBs level is ≥10 IU/L at age 9 months, immunity is proven. If HBsAg is positive, the infant has become infected despite prophylaxis: refer to an appropriate specialist. If HBsAg is negative and anti-HBs level is <10 IU/L at age 9 months, give a further 3 doses of HepB at least 4 weeks apart. Recheck serology 4 weeks after the last dose. If there is no seroconversion after the third further dose of HepB (ie, if anti‑HBs is still <10 IU/L), discuss with a specialist. |
|
All other vaccines should be administered as per the Schedule. |
|
9.5.3. Catch-ups for children and adolescents
9.5.3. Catch-ups for children and adolescents
HepB is recommended and funded for everyone aged under 18 years. If HepB is not given during the first year of life, three doses of vaccine are recommended given 0, 1 and 6 months.
For adolescents aged 10–15 years, an alternative two-dose hepatitis B catch-up schedule may be considered using the monovalent HepB with the second dose given six months after the first.
See Appendix 2 for catch-up schedules.
Children and adolescents with liver or kidney disease
HepB vaccine is funded for liver or kidney transplant patients (recommend six months post-transplant) and for dialysis patients.
See Figure 9.3 and Figure 9.4 for serological testing and vaccination recommendations. If non-immune, children aged under 16 years should receive three doses of HepB; those aged 16 years and older should receive four doses of HepB given at 0, 1, 2 and 12 months. If there is an inadequate immune response to the initial three-dose HepB series (see Figure 9.4), give a further three doses, as appropriate for age.
See also ‘Solid organ transplantation’ in section 4.3.10, ‘Chronic kidney disease’ in section 4.4 and ‘Chronic liver disease’ in section 4.5.
9.5.4. Eligible adults aged 18 years and older
9.5.4. Eligible adults aged 18 years and older
Table 9.6: Hepatitis B vaccine schedules for eligible adults aged 18 years and older
|
Who |
Vaccine |
Dose |
Volume (mL) |
Number of doses |
Schedule |
|---|---|---|---|---|---|
|
Dialysis patients, liver or kidney transplant patients |
HepB |
40 µg |
1.0 (x2) |
3 |
0, 1 and 6 months |
|
HIV patients |
HepB |
20 µg |
1.0 |
4 |
0, 1, 2 and 12 months |
|
Other eligible adults (see Table 9.4) |
HepB |
20 µg |
1.0 |
3 |
0, 1 and 6 months |
| Adult 1 dose (1 mL, 20 micrograms), according to schedule in table above; in adults on dialysis, or who are liver or kidney transplant patients, give two doses at separate sites (1 mL each, total dose 40 micrograms). | |||||
Adult dialysis or adult liver or kidney transplant patients
These adults may have a reduced response to HepB,[36, 42] so three higher doses (40 µg per dose) are recommended and funded.
See section 9.5.7 for information about post-vaccination serology.
(See also ‘Solid organ transplantation’ in section 4.3.10 and recommendations provided in IMAC factsheet ‘Immunisation for adults pre-dialysis, on dialysis or pre-/post-kidney transplant’ available on the IMAC website (external link).)
Adult HIV patients
Adult HIV patients should receive four doses of HepB (20 µg per dose) at 0, 1, 2 and 12 months.
(See also ‘HIV infection’ in section 4.3.12 and recommendations provided in IMAC factsheet Immunisation for adults with HIV infection available on the IMAC website (external link).)
Other eligible adults
The optimal dosing regime is three doses of 20 µg HepB given at 0, 1 and 6 months. See the manufacturer’s data sheet for sub-optimal accelerated HepB schedules if dosing is time constrained. For other eligible adults, see Table 4.8, ‘Other special groups’ in section 4.6.
9.5.5. Pregnancy and breastfeeding
9.5.5. Pregnancy and breastfeeding
HepB may be given during pregnancy and while breastfeeding. Acute HBV infection in pregnant women may result in severe acute hepatitis for the mother, with associated increased risk of fetal loss or neonatal infection. Vaccination should not be withheld from a susceptible pregnant woman at increased risk of acquiring hepatitis B (eg, the sexual partner of an injecting drug user, or known infected male).
9.5.6. (Re)vaccination
9.5.6. (Re)vaccination
Hepatitis B-containing vaccines are funded for vaccination and revaccination of eligible children, as follows. See also sections 4.2 and 4.2.5.
DTaP-IPV-HepB/Hib (Infanrix-hexa)
An additional four doses (as appropriate) of DTaP-IPV-HepB/Hib are funded for vaccination or revaccination of children aged under 10 years:
- post-HSCT or chemotherapy
- pre- or post-splenectomy
- pre- or post-solid organ transplant
- undergoing renal dialysis
- prior to planned or following other severely immunosuppressive regimens.
Up to an additional four doses (as appropriate) of DTaP-IPV-HepB/Hib are funded for children aged 10 years to under 18 years for (re)vaccination of patients post HSCT. This is an off-label use of this vaccine which requires a prescription from an authorised prescriber or the patient’s specialist. In this group, all prior immunity will have been lost and revaccination is equivalent to a primary immunisation schedule. DTaP-IPV-HepB/Hib not recommended for other severely immunocompromised individuals aged 10-18 years that require revaccination as they may have residual immune memory.
Monovalent HepB
HepB is funded for children aged under 18 years who are considered not to have achieved a positive serology and require additional vaccination.
9.5.7. Serological testing
9.5.7. Serological testing
- Serological testing is not routinely recommended – immunisation is highly effective.
- Most people with documented evidence of three HepB vaccinations will be immune for life.
- Unnecessary testing leads to unnecessary extra vaccination.
- Infants born to HBsAg-positive mothers and some individuals who require protection in relation to their employment (eg, health care professionals) require post-vaccination serology.
- Where there is concern about immunity follow Figure 9.3.
Screening for chronic infection
Screening for the antigen (HBsAg) is useful where there is increased likelihood of the individual already being infected.
The Hepatitis Foundation of New Zealand recommends that the following individuals are most at risk of HBV:[43]
- people of Māori, Pacific or Asian ethnicity, unless fully vaccinated with HepB vaccine as an infant
- people born in an area of high hepatitis B endemicity, including Asia, the Pacific Islands, Africa, the Middle East, southern Europe or the northern or eastern parts of New Zealand’s North Island
- people born to a mother or who have a close family member who has chronic HBV infection
- people who live with someone who has HBV
- people who have had unprotected sexual contact with an HBV-infected person
- people who have ever injected drugs
- people who have received a tattoo using unsterile equipment.
Screening for HBsAg is also part of routine antenatal care (see section 9.5.2).
All HBsAg-positive individuals should be offered follow-up under the Hepatitis Foundation Hepatitis B Follow-up Programme to enable early diagnosis and treatment of the complications of severe liver disease and hepatocellular carcinoma. Vaccination is funded for household or sexual contacts of HBsAg-positive people (ie, contacts of people with acute or chronic HBV infection).
To confirm chronic HBV status, repeat testing after 6 months and if still positive, refer patient to The Hepatitis Foundation.
Serological testing for high-risk groups
Serological testing is not routinely recommended – immunisation is highly effective.
- Serological testing is only indicated in high-risk groups (see Table 9.7). These high-risk groups are at higher risk of exposure to HBV, at higher risk of having severe disease or are more susceptible to disease.
- A flow diagram (Figure 9.3) is included to assist in deciding whether pre- and/or post-vaccination serological testing is indicated. Figure 9.3 may be used for any individual aged 12 months or older, such as for the management of blood and body fluid exposures, or when an adult presents to primary care.
The non-responder protocol
Most vaccine recipients will develop a high anti-HBs titre, usually greater than 100 IU/L, which usually wanes over time.
Fully vaccinated individuals (ie, those who have received three documented doses of HepB) who have at any time had anti-HBs antibody titre of ≥10 IU/L do not need any booster doses, even if antibodies subsequently wane to undetectable levels, which occurs in most individuals by seven years after the last vaccination. Adults have been shown to have an anamnestic antibody and cellular immune response to a HepB dose given 20–30 years after the primary immunisation.[24] If exposed to HBV, they will have a secondary anamnestic immune response that will prevent replication of the virus.[1, 44]
Note: Some laboratories may require a higher anti-HBs antibody level for proof of immunity. Please follow the testing laboratory’s interpretative comments.
If a high-risk individual does not achieve a titre of ≥10 IU/L by four weeks following one HepB dose, they should be considered a non-responder and follow the non-responder protocol (Figure 9.4). Where necessary, intradermal administration of a third course enables fewer doses and improved chance of response.[45, 46]
Table 9.7: Individuals at high-risk of hepatitis B infection, for whom serological testing is indicated
Table 9.7: Individuals at high-risk of hepatitis B infection, for whom serological testing is indicated
|
Household or sexual contacts of HBsAg-positive patients (ie, patients with acute or chronic HBV infection) |
|
Current or recent injecting drug users |
|
Individuals who change sexual partners frequently (eg, sex workers) |
|
Immunocompromised individuals, including HIV-positive patients |
|
Following non-consensual sexual intercourse |
|
Individuals prior to planned immunosuppressive therapies for 28 days or more |
|
Individuals following immunosuppressive therapies for 28 days or more |
|
Solid organ and post-HSCT patients |
|
Following percutaneous injury (eg, needle-stick injury) |
|
Adults at occupational-related risk (see section 4.8) |
|
Individuals with haemophilia and other regular recipients of blood products |
|
Inmates of custodial institutions |
|
Individuals with developmental disabilities |
|
People with chronic disease (eg, chronic renal failure requiring haemodialysis, or chronic liver disease) |
|
Migrants from HBV endemic regions (where HBsAg prevalence is ≥2 percent)* |
|
* See the Centers for Disease Control and Prevention website (external link) for countries with an HBsAg prevalence ≥2%. Consider combined Hep A and B vaccination for individuals travelling to these regions. |
Figure 9.3: Flow diagram for serological testing for hepatitis B
Figure 9.3: Flow diagram for serological testing for hepatitis B



|
a. HBIG may be recommended for non-immune individuals. See Table 9.8. b. Do not count any birth doses of HepB vaccine. See Table 9.4 for the list of funded conditions for HepB vaccine. c. See the manufacturer’s data sheet for accelerated HepB schedules. d. Haemodialysis patients need annual testing and boosting if required. |
Figure 9.4: The non-responder protocol
Figure 9.4: The non-responder protocol
Individual is high-risk (see Table 9.7), has received three documented doses of HepB, and after a booster dose (as indicated in Figure 9.3) continues to have an anti-HBs antibody titre <10 IU/L:
- Check serology for HBsAg and anti-HBc antibody to exclude chronic infection.
- Complete a second course with two further HepB vaccine doses, given at 1 and 6 months after booster dose.
- Repeat the serology four weeks after the final HepB vaccine dose
- If anti-HBs ≥10 IU/L, assume immunity. No further action is required.
- Where there is expertise available to administer intradermal injections, deliver 0.25 ml (5 µg) of HepB intradermally at four sites as tolerated (e.g. each deltoid and anterior thigh) ensuring a pale wheal is formed on the skin.
- Repeat serology four weeks later.
- If anti-HBs remains at ≤10 IU/L, repeat intradermal doses.
- Repeat serology 4 weeks later.
- If, after a second intramuscular three-dose course of HepB followed by two intradermal HepB doses given across multiple injection-sites, the individual has not achieved anti-HBs ≥10 IU/L, they should be considered a persistent non-responder to vaccination.
Persistent non-responders with no immunocompromise who have completed the primary series and further courses of three vaccine doses should be monitored for wild-type disease, but literature reports disease from vaccine failures are rare. They should be considered ‘unprotected’ against hepatitis B and advised to minimise the chance of exposures. Parenteral or mucosal exposure to HBV requires HBIG within 72 hours.
9.6. Contraindications and precautions
See also section 2.1.3 for pre-vaccination screening guidelines and section 2.1.4 for general contraindications for all vaccines.
The only specific contraindication to HepB is anaphylaxis following a previous dose, or individuals with a history of allergic reactions to yeast or any of the vaccine’s components. Immunisation of previously infected subjects is wasteful, but not harmful.
See section 16.6 for contraindications and precautions to DTaP‑IPV‑HepB/Hib vaccine.
9.7. Potential responses and AEFIs
See section 16.7 for potential responses and AEFIs with DTaP‑IPV‑HepB/Hib vaccine.
9.7.1. Potential responses
9.7.1. Potential responses
Minor side-effects – including local tenderness and redness, nausea, diarrhoea, general malaise and fever – are more common in adults than in children and, except for local reactions, occur at rates close to those seen with a placebo. Minor reactions reported after receiving the vaccine include a temperature >37.7°C in 1–6 percent; pain in 3–29 percent; and erythema, headache or swelling in 3 percent of vaccine recipients.
9.7.2. AEFIs
9.7.2. AEFIs
Allergic reactions have been reported but are rare. Anaphylaxis following vaccination is extremely rare (estimated to be 1.1 cases per million doses).[47]
A number of studies have examined and failed to find disease events linked to hepatitis B immunisation.[48] These studies have documented no increased risk of multiple sclerosis,[49, 50, 51] diabetes, chronic fatigue syndrome,[52] encephalomyelitis or hair loss.[53] Rarely, transient thrombocytopenia[54] and myalgia and arthralgia[55, 56] have been reported after HepB vaccination.
9.8. Public health measures
It is a legal requirement that all cases of acute hepatitis B infection be notified to the local medical officer of health.
The elimination of HBV transmission is now a realistic public health goal[7, 57] especially with the proven effectiveness and safety record of HepB.[58] Achievement of this goal is being facilitated by the implementation of triple elimination strategies in the Western Pacific Region to prevent mother-to-child transmission of HIV, HBV and syphilis,[8] and the use of birth dosing HepB regimes, either universally or as directed.
It is important to ensure vaccination programmes are maintained for the at-risk populations, especially babies of mothers with chronic hepatitis B infection.
Babies born to HBsAg-positive mothers should be notified at birth. The prevention of perinatal transmission is covered in section 9.5.2.
For further information about public health measures, see the Hepatitis B chapter of the Communicable Disease Control Manual.
9.8.1. Passive immunisation
9.8.1. Passive immunisation
HBIG is prepared from donated blood plasma and contains high levels of anti-HBs antibody (see Appendix 6). It is given after exposure to HBV and provides passive anti-HBs antibody protection against acute and chronic HBV disease. HBIG prophylaxis should be given in combination with the HepB to confer both passive and active immunity after exposure.
The efficacy of HBIG alone in preventing clinical hepatitis B infection is about 75 percent in adults, but the protection lasts only for a few months.[1]
Whenever immediate protection is required, immunisation with a vaccine should be combined with simultaneous administration of HBIG at a different site. It has been shown that passive immunisation with HBIG does not suppress the active immune response to vaccination. A single dose of HBIG is sufficient (usually 400 IU for adults, 100–110 IU for newborns). If infection has already occurred at the time of the first immunisation, virus replication is unlikely to be inhibited completely, but severe illness and, more importantly, the development of chronic HBV infection may be prevented, particularly in the infants of HBsAg-positive mothers.
The management of contacts is summarised in Table 9.8.
Table 9.8: Management of contacts of hepatitis B cases
|
Contact |
Serological testing of contact |
Immunoglobulin |
Immunisation |
|---|---|---|---|
|
Any sexual contact, including protected sex |
Yes |
Yes, immediately after blood taken |
Yes, immediately after blood taken |
|
Household, mucosal or percutaneous |
Yes |
Yes, if serology negative |
Yes, if serology negative |
|
Other |
Yes |
No |
Yes, if serology negative |
| Source: Health New Zealand | Te Whatu Ora. Communicable Disease Control Manual. Wellington: Health NZ (accessed 24 July 2024). | |||
For further definitions and details see the Communicable Disease Control Manual.
9.9. Variations from the vaccine data sheet
See section 16.9 for variations from the DTaP-IPV-HepB/Hib (Infanrix‑hexa) data sheet.
Health New Zealand advises that two doses Engerix-B 20 µg, given six months apart, may be given to adolescents aged 10–15 years. The manufacturer’s data sheet recommends three doses of 10 µg given at 0, 1 and 6 months, but in circumstances where compliance may not be assured, giving 20 µg per dose increases the proportion of recipients protected after the first and second doses.
Although Health New Zealand and the data sheet recommend 0.5 ml Engerix-B 10 µg (paediatric formulation) for neonates born to HBV infected mothers, where the paediatric presentation is unavailable, the data sheet advises that Engerix-B 20 µg can be given to children from birth up to the age of 10 years.
Health New Zealand recommends giving three doses of 40 ug HepB (ie, two doses of Engerix-B 20 µg per visit) to be given 0, 1, and 6 months for adult renal dialysis patients, liver or kidney transplant patients. For adults with HIV, four doses of Engerix-B 20 µg is recommended given at 0, 1, 2 and 12 months. The data sheet advises four doses of 40 µg (ie, two doses for Engerix-B 20 µg per visit) given at 0, 1, 2 and 6 months for chronic haemodialysis patients and other individuals who have an impairment of their immune system.
References
References
References
- Van Damme P, Ward J, Shouval D,Zanetti P. 2018. Hepatitis B Vaccines, in Plotkin's Vaccines (7th edition), Plotkin S, Orenstein W, Offit P, and Edwards K (eds). Elsevier: Philadelphia, US.
- McMahon BJ, Alward WL, Hall DB, et al. Acute hepatitis B virus infection: relation of age to the clinical expression of disease and subsequent development of the carrier state. Journal of Infectious Diseases, 1985. 151(4): p. 599-603.
- Stockdale AJ, Kreuels B, Henrion MYR, et al. The global prevalence of hepatitis D virus infection: systematic review and meta-analysis. Journal of Hepatology, 2020.
- Bond WW, Favero MS, Petersen NJ, et al. Survival of hepatitis B virus after drying and storage for one week. Lancet, 1981. 1(8219): p. 550-1.
- Alter HJ. To have B or not to have B: vaccine and the potential eradication of hepatitis B. Journal of Hepatology, 2012. 57(4): p. 715-7.
- Papastergiou V, Lombardi R, MacDonald D,Tsochatzis EA. Global epidemiology of hepatitis B Virus (HBV) infection. Current Hepatology Reports, 2015. 14(3): p. 171-178.
- World Health Organization. 2016. WHO Global Health Sector Strategy on Viral Hepatitis 2016–2021 (ed.), Geneva, Switzerland: World Health Organization. URL: https://apps.who.int/iris/handle/10665/246177 (external link) (accessed 10 May 2022)
- World Health Organization. Hepatitis B vaccines: WHO position paper - July 2017. Weekly Epidemiological Record, 2017. 92(27): p. 369-392.
- World Health Organization. China steers towards zero new hepatitis B infections (Press release). World Health Organization. 30 March 2019 URL: https://www.who.int/hepatitis/news-events/china-hbv-childhood-vaccination/en/ (external link). (accessed 3 July 2020)
- Chiang CJ, Yang YW, You SL, et al. Thirty-year outcomes of the national hepatitis B immunization program in Taiwan. JAMA, 2013. 310(9): p. 974-6.
- Harris AM. 2020. Hepatitis B. in CDC 2020 Yellow Book. Health Information for International Travel. New York, New York. URL: https://wwwnc.cdc.gov/travel/yellowbook/2020/travel-related-infectious-diseases/hepatitis-b (external link). (accessed 3 July 2020)
- Ott JJ, Stevens GA, Groeger J,Wiersma ST. Global epidemiology of hepatitis B virus infection: new estimates of age-specific HBsAg seroprevalence and endemicity. Vaccine, 2012. 30(12): p. 2212-9.
- Milne A, Allwood GK, Moyes CD, et al. Prevalence of hepatitis B infections in a multiracial New Zealand community. New Zealand Medical Journal, 1985. 98(782): p. 529-32.
- Moyes C ,Milne A. Hepatitis B markers in 14–15 year olds in the Bay of Plenty. The New Zealand Medical Journal, 1986. 99(809): p. 662–4.
- Rainger W, Solomon N ,Jones N. Immunisation coverage and risk factors for immunisation failure in Auckland and Northland. New Zealand Public Health Report, 1998. 5(7): p. 49–51.
- Ramadas D, Moyes CD ,Ramadas G. Immunisation status of children in the eastern Bay of Plenty. New Zealand Medical Journal, 1992. 105(942): p. 378-9.
- Stehr-Green P, Briasco C ,Baker M. How well are we protecting our children? An immunisation coverage survey in Hawke’s Bay. The New Zealand Medical Journal, 1992. 105(938): p. 277–9.
- The Hepatitis Foundation of New Zealand. 2030 Targets. The Hepatitis Foundation of New Zealand,; URL: https://www.hepatitisfoundation.org.nz/2030-targets (external link). (accessed 11 May 2020)
- Robinson T, Bullen C, Humphries W, et al. The New Zealand Hepatitis B Screening Programme: screening coverage and prevalence of chronic hepatitis B infection. New Zealand Medical Journal, 2005. 118(1211): p. U1345.
- Mann J ,Roberts M. Modelling the epidemiology of hepatitis B in New Zealand. J Theor Biol, 2011. 269(1): p. 266-72.
- Addidle M. Impact of universal hepatitis B vaccination on antenatal hepatitis B prevalence in the Midlands region of the North Island, New Zealand. New Zealand Medical Journal, 2011. 124(1332): p. 40-4.
- Lim TH, Gane E, Moyes C, et al. Serological and clinical outcomes of horizontally transmitted chronic hepatitis B infection in New Zealand Maori: results from a 28-year follow-up study. Gut, 2015. 64(6): p. 966-72.
- Lee C, Gong Y, Brok J, et al. Effect of hepatitis B immunisation in newborn infants of mothers positive for hepatitis B surface antigen: systematic review and meta-analysis. BMJ, 2006. 332(7537): p. 328-36.
- Van Damme P, Dionne M, Leroux-Roels G, et al. Persistence of HBsAg-specific antibodies and immune memory two to three decades after hepatitis B vaccination in adults. Journal of Viral Hepatitis, 2019. 26(9): p. 1066-1075.
- Gruber C, Warner J, Hill D, et al. Early atopic disease and early childhood immunization--is there a link? Allergy, 2008. 63(11): p. 1464-72.
- Moyes CD, Milne A ,Waldon J. Very low dose hepatitis B vaccination in the newborn: anamnestic response to booster at four years. Journal of Medical Virology, 1990. 30(3): p. 216-8.
- West DJ ,Calandra GB. Vaccine induced immunologic memory for hepatitis B surface antigen: implications for policy on booster vaccination. Vaccine, 1996. 14(11): p. 1019-27.
- Van Der Meeren O, Bleckmann G ,Crasta PD. Immune memory to hepatitis B persists in children aged 7-8 years, who were vaccinated in infancy with 4 doses of hexavalent DTPa-HBV-IPV/Hib (Infanrix hexa) vaccine. Human Vaccines & Immunotherapeutics, 2014. 10(6): p. 1682-7.
- Su WJ, Liu CC, Liu DP, et al. Effect of age on the incidence of acute hepatitis B after 25 years of a universal newborn hepatitis B immunization program in Taiwan. Journal of Infectious Diseases, 2012. 205(5): p. 757-62.
- McMahon BJ, Bulkow LR, Singleton RJ, et al. Elimination of hepatocellular carcinoma and acute hepatitis B in children 25 years after a hepatitis B newborn and catch-up immunization program. Hepatology, 2011. 54(3): p. 801-7.
- Perz JF, Elm JL, Jr., Fiore AE, et al. Near elimination of hepatitis B virus infections among Hawaii elementary school children after universal infant hepatitis B vaccination. Pediatrics, 2006. 118(4): p. 1403-8.
- Chen D-S. Hepatitis B vaccination: The key towards elimination and eradication of hepatitis B. Journal of Hepatology, 2009. 50(4): p. 805-16.
- Chang M-H. 2011. Hepatitis B virus and cancer prevention, in Clinical Cancer Prevention, Senn H-J,Otto F (eds). Springer: Berlin & Heidelberg.
- Lee CL ,Ko YC. Hepatitis B vaccination and hepatocellular carcinoma in Taiwan. Pediatrics, 1997. 99(3): p. 351-3.
- Whitford K, Liu B, Micallef J, et al. Long-term impact of infant immunization on hepatitis B prevalence: a systematic review and meta-analysis. Bulletin of the World Health Organization, 2018. 96(7): p. 484-497.
- el-Reshaid K, al-Mufti S, Johny KV,Sugathan TN. Comparison of two immunization schedules with recombinant hepatitis B vaccine and natural immunity acquired by hepatitis B infection in dialysis patients. Vaccine, 1994. 12(3): p. 223-34.
- Committee on Infectious Diseases. Update on timing of hepatitis B vaccination for premature infants and for children with lapsed immunisation. Pediatrics, 1994. 94(3): p. 403–4.
- American Academy of Pediatrics. 2018. Hepatitis B. in Red Book: 2018 Report of the Committee on Infectious Diseases, Kimberlin D, Brady M, Jackson M, and Long S (eds). URL: https://redbook.solutions.aap.org/redbook.aspx (external link). (accessed 3 July 2020)
- Scheller NM, Svanstrom H, Pasternak B, et al. Quadrivalent HPV vaccination and risk of multiple sclerosis and other demyelinating diseases of the central nervous system. JAMA, 2015. 313(1): p. 54-61.
- Benarroch EE. Postural tachycardia syndrome: a heterogeneous and multifactorial disorder. Mayo Clinic Proceedings, 2012. 87(12): p. 1214-25.
- Borucki AN ,Greco CD. An update on complex regional pain syndromes in children and adolescents. Current Opinion in Pediatrics, 2015. 27(4): p. 448-52.
- Roukens AH ,Visser LG. Hepatitis B vaccination strategy in vaccine low and non-responders: a matter of quantity of quality? Hum Vaccin, 2011. 7(6): p. 654-7.
- Hepatitis Foundation of New Zealand. Hepatitis B for health professionals. URL: https://www.hepatitisfoundation.org.nz/health-professionals/hepatitis-b-health-professionals (external link). (accessed 20 January 2020)
- European Consensus Group on Hepatitis B Immunity. Are booster immunisations needed for lifelong hepatitis B immunity? European Consensus Group on Hepatitis B Immunity. Lancet, 2000. 355(9203): p. 561-5.
- Yousaf F, Gandham S, Galler M, et al. Systematic review of the efficacy and safety of intradermal versus intramuscular hepatitis B vaccination in end-stage renal disease population unresponsive to primary vaccination series. Renal Failure, 2015. 37(7): p. 1080-8.
- David MC, Ha SH, Paynter S,Lau C. A systematic review and meta-analysis of management options for adults who respond poorly to hepatitis B vaccination. Vaccine, 2015. 33(48): p. 6564-9.
- Bohlke K, Davis RL, Marcy SM, et al. Risk of anaphylaxis after vaccination of children and adolescents. Pediatrics, 2003. 112(4): p. 815-20.
- Institute of Medicine: Committee to Review Adverse Effects of Vaccines. 2012. Adverse effects of vaccines: Evidence and causality (ed.), Washington, DC: The National Academies Press. URL: https://www.nap.edu/catalog/13164/adverse-effects-of-vaccines-evidence-and-causality (external link) (accessed January 2020)
- Expanded Programme on Immunization (EPI). Expanded programme on immunization (EPI). Lack of evidence that hepatitis B vaccine causes multiple sclerosis. Weekly Epidemiological Record, 1997. 72(21): p. 149-52.
- Mouchet J ,Begaud B. Hepatitis B vaccination and central demyelination - History, description and observed/expected analyses of 624 cases reported to the French pharmacovigilance over a 20-year period. Vaccine, 2019. 37(15): p. 2142-2148.
- Sadovnick AD ,Scheifele DW. School-based hepatitis B vaccination programme and adolescent multiple sclerosis. The Lancet, 2000. 355(9203): p. 549-50.
- Health and Welfare Canada. Report of the working group on the possible relationship between hepatitis B vaccination and the chronic fatigue syndrome. Canadian Communicable Disease Report, 1993. 19(4): p. 25–8.
- Wise RP, Kiminyo KP ,Salive ME. Hair loss after routine immunizations. JAMA, 1997. 278(14): p. 1176-8.
- Ronchi F, Cecchi P, Falcioni F, et al. Thrombocytopenic purpura as adverse reaction to recombinant hepatitis B vaccine. Archives of Disease in Childhood, 1998. 78(3): p. 273-4.
- Fisher MA, Eklund SA, James SA,Lin X. Adverse events associated with hepatitis B vaccine in U.S. children less than six years of age, 1993 and 1994. Annals of Epidemiology, 2001. 11(1): p. 13-21.
- McMahon BJ, Helminiak C, Wainwright RB, et al. Frequency of adverse reactions to hepatitis B vaccine in 43,618 persons. American Journal of Medicine, 1992. 92(3): p. 254-6.
- Ni YH, Chang MH, Wu JF, et al. Minimization of hepatitis B infection by a 25-year universal vaccination program. Journal of Hepatology, 2012. 57(4): p. 730-5.
- Romanò L, Paladini S, Van Damme P,Zanetti AR. The worldwide impact of vaccination on the control and protection of viral hepatitis B. Digestive and Liver Disease, 2011. 43 Suppl 1(Suppl 1): p. S2-7.